软件
Allele-Specific拷贝数分析肿瘤(ASCAT)
ASCAT方法获得拷贝数的肿瘤细胞,正常细胞占外加剂和肿瘤的非整倍性。纯度ASCAT推断肿瘤(肿瘤细胞的比例)和倍性(每个肿瘤细胞DNA的数量,表示为单倍体基因组)的倍数从SNP数组或大规模并行测序数据,并计算出全基因组allele-specific拷贝数概要文件(副本的数量两个亲本的等位基因的SNP位点在基因组)。
最新的ASCAT版本可在GitHub上作为R包,:https://github.com/VanLoo-lab/ascat。指导安装和试着在GitHub页面提供的软件。
运行ASCAT
在其最简单的形式(与正常数据,没有GC波校正和所有女性)样品,ASCAT可以运行如下:
库(ASCAT)
ascat。公元前= ascat.loadData (“Tumor_LogR。txt”、“Tumor_BAF。txt”、“Germline_LogR。txt”、“Germline_BAF.txt”)
ascat.plotRawData (ascat.bc)
ascat。公元前= ascat.aspcf (ascat.bc)
ascat.plotSegmentedData (ascat.bc)
ascat。输出= ascat.runAscat (ascat.bc)
ascat。loadData函数默认情况下假设所有样本都是女性。一个额外的可选参数(性别=…)允许设置的性别样本(在矢量格式,使用“XX”,雌性和雄性“XY”)。
ASCAT可以运行在不同的模式:没有匹配的正常数据,与logR校正(GC含量和复制时间),multi-segmentation和高通量测序(高温超导)数据。示例运行ASCAT可以找到使用这些不同的模式在这里。
输入数据格式和支持平台
1)SNP数组
ASCAT平台和与对象无关,适用于Illumina公司和Affymetrix SNP数组。所需的输入包括矩阵LogR和B等位基因频率(BAF)数据(行探针或SNP位点和列样品)。ASCAT需要相同的格式化LogR和肿瘤和生殖系BAF文件数据(匹配样本在所有四个文件)匹配的行。精确的数据格式的例子,请参阅我们的模拟的示例数据(7.62 MB,邮编)。
ASCAT输入数据可以直接从Illumina公司GenomeStudio或者可以获得来自Affymetrix玻璃纸文件,例如通过PennCNV库。我们使用的管道(推荐)Affymetrix SNP 6.0数组可以在GitHub上的R包。
请注意,你需要两个适应文件对于这个管道,一个包含SNP AffySNP6平台的位置(12.68 MB,邮编)和一个基因型集群文件(33.25 MB,邮编)这是编译的一系列约5000验证正常样本。
2)高温超导数据
对于高温超导数据,ASCAT需要BAM文件和引用文件(在GitHub上市页)所以它可以获得等位基因数和派生logR和BAF值。后logR / BAF文件生成(ascat.prepareHTS),可以使用另一个ASCAT函数来执行所有的标准步骤(加载logR / BAF,纠正logR反是,分段跟踪和CNA概要文件)。
有针对性的测序数据,我们实现了一个定制函数,识别高质量的单核苷酸多态性(ascat.prepareTargetedSeq)进行调查。这一步必须完成一批正常样品(没有肿瘤样本)和生成logR和BAF前值。更多的信息关于如何获得高温超导CNA概要数据可以发现我们的GitHub页面。
3)附加信息
ASCAT也可以运行在其他物种的数据,例如,从犬乳腺癌SNP数组或从外显斑马鱼黑色素瘤。方法利用SNP位点,但是不是单倍体或纯合子(天生的)物种(例如近交品系小鼠)。
异形通过SNP数组或大规模并行测序样品往往受到“波构件”,在一定程度上与周边地区的GC含量(如。本文通过迪斯et al。)。我们实现了一个GC ASCAT波校正,并推荐向管道中添加这一步如果输入数据没有通过替代logR校正的方法。我们最初的GC校正方法(ASCAT 2.2)是基于一个最初实现的程et al .,基因组生物学12:R80, 2011年。我们已经延长这种校正方法正确的GC含量和复制时间(从3.0版)。
一个重要平台和normalization-specific参数是函数内的归一化参数(γ)ascat.runAscat。这个参数代表LogR下降的变化从两份一份细胞的100%。对于大规模并行测序数据,伽马应该设置为1。数组数据,由于数组背景信号和定制数组规范化程序,γ往往在实践中明显降低。0.55的默认设置为许多工作但不是所有SNP数组(例如Illumina公司109 k数组作为加工通过BeadStudio / GenomeStudio和Affymetrix SNP 6.0数组处理通过PennCNV库)。其他SNP阵列平台(和规范化程序),我们建议检查γ的值通过比较男性和女性生殖系样本(LogR值评估不同性别之间的X染色体探针,相对于其它的基因组),或通过一个X染色体滴定系列。
ASCAT输出
ASCAT的输出,以及如何解释它,描述这本书一章。
遗留版本和数据
历史版本的ASCAT可用的一部分我们的GitHub版本。我们建议总是使用最新版本,但我们提供历史版本遗留的原因。
重大变化在原始ASCAT 1.0版本是:
●可用性是一个易于使用的和连贯的R软件套件(2.0)
●主要提高运算速度(2.0)
●平台独立性(2.0)
●更新的核心算法更好的性能和结果(2.0和2.2)
●增加生殖系基因型预测,从而扩展无与伦比的肿瘤样本(2.0)
●适应ASPCF分割算法来提高灵敏度和低噪声样本和增加鲁棒性噪声样本(2.1)
●添加性别参数,允许正确处理拷贝数的X染色体畸变男性样本(2.2)
●增加GC修正代码(2.2)
●适应允许手动改装的样本(2.3)
●适应和添加到输出数据结构(2.3)
●可用性作为R包(2.4)
●添加多试样细分为样本,预计分享断点(2.5)
●添加定制的方法从高温超导数据生成logR和BAF (3.0)
●增加有针对性的测序数据的预处理步骤(3.1)
乳腺癌SNP数组的数据我们最初ASCAT出版也可用。数据由LogR和肿瘤和生殖系SNP BAF值数组的数据。我们还包括肿瘤LogR GC偏见的数据调整后使用中描述的方法迪斯et al .,核酸研究,36:e126, 2008188bet体育网址。由于隐私法规,数据是密码保护。请联系我们获得访问。
一个脚本用于分析这些Illumina公司109 k乳腺癌SNP阵列数据使用在GitHub ASCAT 1.0是可用。
Subclonal拷贝数分析:巴腾堡蛋糕算法
化验subclonal拷贝数变化在大规模并行测序数据中,我们创建了巴腾堡蛋糕算法,基于底层ASCAT原理和方程和单体型逐步1000基因组SNP位点。的巴腾堡蛋糕算法最初是这里所描述的,现在是可用的GitHub。
常见问题(FAQ)
我可以用ASCAT(生殖系)CNV分析?
ASCAT是一个工具来检测体细胞拷贝数改变(CNAs)癌症样本不能用于检测生殖系拷贝数变异基因拷贝数异变。CNV指生殖系的一个变种,在人口多态。为了避免混淆,对于体细胞拷贝数改变肿瘤样本,我们推荐使用术语CNA总是。
我应该使用哪个版本的ASCAT ?
我们建议ASCAT总是使用最新版本。
可以应用于细胞系ASCAT吗?
ASCAT将数据匹配的细胞系。然而,它并不适合分析无与伦比的细胞系数据,作为生殖系基因型预测工具利用了信号混在正常细胞来推断生殖系基因型。因为大多数细胞系在实践中是无与伦比的,ASCAT很可能不是一种理想的细胞系的分析数据的方法。
我什么时候应该使用ASCAT ?我什么时候应该使用巴腾堡蛋糕?
巴腾堡蛋糕算法是专门为检测subclonal拷贝数变化对全基因组测序数据。当前版本的巴腾堡蛋糕也可以推断出纯洁和倍性的数据,并将我们的方法选择全基因组测序数据的分析。
为其他测序数据的分析(外显子组或目标下拉),单体型逐步有更多增值有限,我们建议使用ASCAT。ASCAT还支持从其他物种的分析数据并分析多种SNP数组。
关于
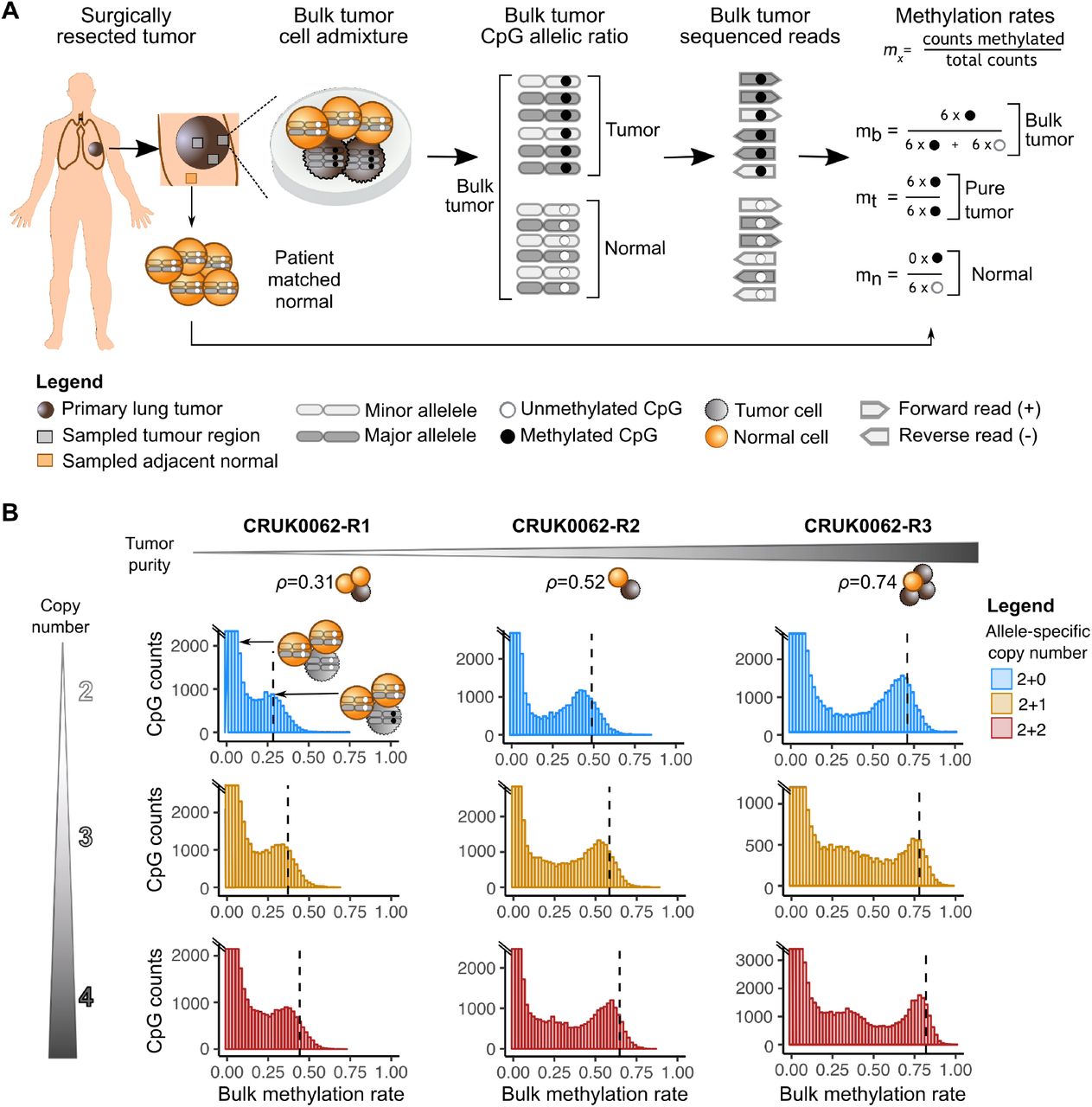
CAMDAC是deconvolving大部分肿瘤的方法重亚硫酸盐测序数据。大部分肿瘤DNA甲基化是由正常细胞外加剂抱愧蒙羞和可变肿瘤DNA内容由拷贝数变化。CAMDAC推断肿瘤纯度、倍性和从散装methylomes allele-specific拷贝数州,结合了这些量和参考大量正常的甲基化分数在每个站点上,肿瘤,纯每CpG甲基化分数的网站。我们表明,相邻patient-matched邻近正常组织是一个合适的代理正常浸润的细胞群。CAMDAC-derived methylomes代表平均每CpG甲基化等位基因在肿瘤人群和权力真正的发现肿瘤差异甲基化事件。
资源
- - - - - -CAMDAC github库
——CAMDAC文档(rrb/WGBS)
- - - - - -CAMDAC预印本
引用
如果你在你的工作中使用CAMDAC,请列举:
女子拉上磨蹭很久,E。曼沙,N.E.Castignani C。Tanić,M。威尔逊,G.A.Dietzen, M。Dhami, P。Vaikkinen, H。Verfaillie,。Cotobal马丁,C。贝克,T。沃特金斯,T.B.K.Selvaraju, V。Jamal-Hanjani, M。,卡奴,N。印度,N。2月,。TRACERx财团Swanton, C。贝克,S。, Demeulemeester, J., and Van Loo, P. 2022. Copy number-aware deconvolution of tumor-normal DNA methylation profiles. bioRxiv, doi: https://doi.org/10.1101/2020.11.03.366252.
CAMDAC
成对tumor-normal研究设计,CAMDAC接受通过allele-counting BAM文件和收益,拷贝数的召唤,反褶积和微分甲基化测试。例如:
' ' '
图书馆(CAMDAC)
肿瘤=create_camdac_sample(“P1_T1”, bam_file =“tumor.bam”)
正常=create_camdac_sample(“P1_N1”, bam_file =“normal.bam”)
配置=create_camdac_config(outdir = "。/results", bsseq="wgbs", bsseq_lib="pe", build="hg38")
pipeline_tumor_normal(肿瘤,正常配置)
' ' '
CAMDAC结果
为每一个管道阶段CAMDAC返回多个输出文件。Deconvolved甲基化率R数据帧存储在对象“CAMDAC_results_per_CpG.RData”。
重要字段包括:
•m_t: CAMDAC纯肿瘤甲基化率
•m_t_low: CAMDAC纯肿瘤低甲基化率HDI99边界
•m_t_high: CAMDAC纯肿瘤甲基化率HDI99上界
•问题:Tumour-normal DMP概率
•CG_CN: CpG等位基因拷贝数
•nA:主要的等位基因拷贝数
•注:小等位基因拷贝数
•段:拷贝数段的端点
支持的平台上
CAMDAC目前支持两种平台,减少表示亚硫酸氢酸性亚硫酸盐测序(rrb)和全基因组测序(WGBS)。CAMDAC验证了这些平台使用俾斯麦对准器。
常见问题
我在哪里可以找到CAMDAC输出的信息吗?
年底CAMDAC输出文件描述的文档(rrb / WGBS)各自的平台。
我可以用CAMDAC没有匹配的正常组织吗?
是的,CAMDAC可以通过池运行正常组织不匹配性,从患者样本配对甲基化,在理想的情况下使用相同的图书馆准备协议和预处理管道。
我可以用CAMDAC转移样品吗?
是的,CAMDAC可以应用于转移性肿瘤,然而重要的是要提供一个正常的甲基化剖面的浸润细胞起源的细胞。
最好的方法是什么运行CAMDAC多区样品吗?
对于多区样本,CAMDAC可以使用相同的patient-matched正常组织为每个大部分肿瘤样本参考。
图片来源:
图1 -从CAMDAC纸,可用:https://www.biorxiv.org/content/biorxiv/early/2020/11/04/2020.11.03.366252/F1.large.jpg?width=800&height=600&carousel=1
——CAMDAC QC输出示例(来自rrb手册):https://htmlpreview.github.io/?https: / /github.com/VanLoo-lab/CAMDAC/blob/main/CAMDAC_manual.html
{kind=link}